ЖИ көмекші
ЖИ көмекші


0 / 1

Материалға шағымдану
Бұл материал сайт қолданушысы жариялаған. Материалдың ішінде жазылған барлық ақпаратқа жауапкершілікті жариялаған қолданушы жауап береді. Ұстаз тілегі тек ақпаратты таратуға қолдау көрсетеді. Егер материал сіздің авторлық құқығыңызды бұзған болса немесе басқа да себептермен сайттан өшіру керек деп ойласаңыз осында жазыңыз
Жариялаған:
Талғатова Айсұлу Даулетқызы 13 Желтоқсан 2020
13 Желтоқсан 2020 506
506Шағым жылдам қаралу үшін барынша толық ақпарат жіберіңіз

1 бонус = 1 теңге
Бонусты сайттағы қызметтерге жұмсай аласыз. Мысалы келесі материалды жеңілдікпен алуға болады
Бонусты жинап картаңызға (kaspi Gold, Halyk bank) шығарып аласыз
Бонусты жинап картаңызға (kaspi Gold, Halyk bank) шығарып аласыз
Түсінікті
Материалдар / STUDY of the mechanism of CO TRANSFORMATION ON a Zn-Cu-Al CATALYST USING QUANTUM CHEMICAL CALCULATION
Педагогтарға арналған 400 000-астам
оқу-әдістемелік материалдарды
1 жыл бойы тегін жүктеу мүмкіндігіне ие болыңыз!
1 жыл бойы тегін жүктеу мүмкіндігіне ие болыңыз!
ҚМЖ
Презентациялар
Ашық сабақтар
Тәрбие сабақтар
Көрнекіліктер
Дидактикалық ойындар
Әдіс-тәсілдер
Авторлық бағдарламалар

-96%
Бүгін алсаңыз
жеңілдік
жеңілдік
00
04
57
Уақытыңыз шектеулі
Толығырақ
Барлық 400 000 материалдарды 1 жыл бойы тегін жүктей аласыз
STUDY of the mechanism of CO TRANSFORMATION ON a Zn-Cu-Al CATALYST USING QUANTUM CHEMICAL CALCULATION
Материал туралы қысқаша түсінік
STUDY of the mechanism of CO TRANSFORMATION ON a Zn-Cu-Al CATALYST USING QUANTUM CHEMICAL CALCULATION

Бұл бетте материалдың қысқаша нұсқасы ұсынылған. Материалдың толық нұсқасын жүктеп алып, көруге болады
STUDY of the mechanism of CO TRANSFORMATION ON a Zn-Cu-Al
CATALYST USING QUANTUM CHEMICAL CALCULATION
METHODS
Talgatova Aisulu Dauletovna
Zhetysu University named after I. Zhansugurov
Email: td.ai@mail.ru
The main approaches to considering the mechanism of CO adsorption on low-temperature Zn-Cu-Al catalysts for methanol synthesis are investigated. Using quantum "chemical methods for the calculation of the analysis of the synthesis mechanism, taking into account obra" the use of positively charged chemisorbed complex. The structures of active centers of catalysts are studied. The binding energies between the adsorbed CO molecule and the active center of the catalyst are calculated.
Keywords: Methanol, surface synthesis mechanism, quantum chemical calculation methods, active center, adsorption, binding energy.
Methanol is one of the most widely used chemical products in the world, used in the oil and gas industry as an inhibitor of hydrate formation in the production and transportation of oil and gas. In addition, methanol is used to produce chemical products that are processed into polymer materials, dyes, solvents, medicines and other substances. Methanol by its physical and chemical properties makes it possible to use it not only directly as a fuel [1]. In modern installations, methanol synthesis is carried out using two types of catalysts: high – temperature-zinc – chromium and low-temperature-zinc-copper. Zinc-chromium catalysts are thermally more stable, but have less activity compared to low-temperature zinc-copper catalysts. When using highly active Zn-Cu-Al catalytic systems, the selectivity of methanol synthesis decreases in comparison with Zn-Cr catalysts. However, the service life and thermal stability of Zn-Cu-Al catalysts is significantly higher [2]. Therefore, there is a need to preserve selectivity and create conditions for inhibiting the side process of hydrocarbon synthesis by the Fischer-Tropsch reaction on the metal centers of the catalyst formed under the conditions of thermal decomposition of Zn-Cu clusters. The description of this process using various physical and chemical approaches allows us to take into account the mechanism of formation of the active center on the surface of the Zn-Cu-Al catalyst and the modes of cluster activation. Using the method of mathematical modeling also allows us to solve the problem of testing catalysts of various brands for the synthesis of methanol. Therefore, the main goal of this work is to substantiate the mechanism of methanol synthesis on the surface of Zn-Cu-Al catalysts using quantum chemical calculations.Using the results obtained, it is planned to create a mathematical model of the methanol synthesis process, and the obtained thermodynamic values will be used to find the constants of the reaction rates occurring on the surface of the catalyst. The mathematical model developed in this way will solve the main issues of existing industrial methanol synthesis plants: forecasting the operation of the plant and testing of catalysts from various manufacturers. When drawing up a mathematical model of the process, it was decided to rely on the generally accepted multi-level scheme for constructing the structure of mathematical models of chemical processes, proposed by M. G. Slinko [3]. At the first stage, the mechanism of the process is clarified, on the basis of which a kinetic model of the process and a model of substance transfer in the catalyst layer are compiled. The next step is to create a mathematical model of the reactor and the technological scheme as a whole. Currently, the scientific literature considers two variants of elementary mechanisms that occur in the synthesis of methanol on Zn-Cu-Al catalysts. The first approach is based on the fact that the synthesis of methanol is preceded by reactions of shock substitution of water with carbon dioxide [4]. The water molecule adsorbed at the first stage of the mechanism is subsequently replaced on the surface of the catalyst by a CO2 molecule coming from the gas phase, while the water molecule passes into the gas phase. Then the adsorbed CO2 interacts with hydrogen, resulting in the final product – methanol. This mechanism is characterized by the following scheme of transformation of basic substances, where [Me] is the metal center on the surface of the catalyst:
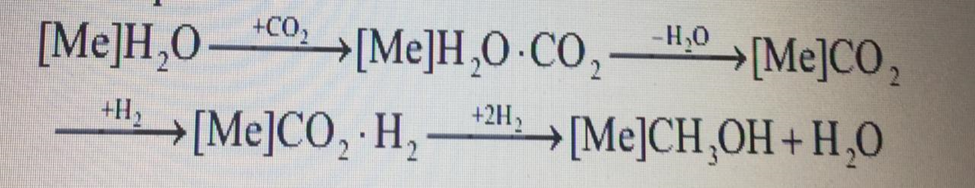
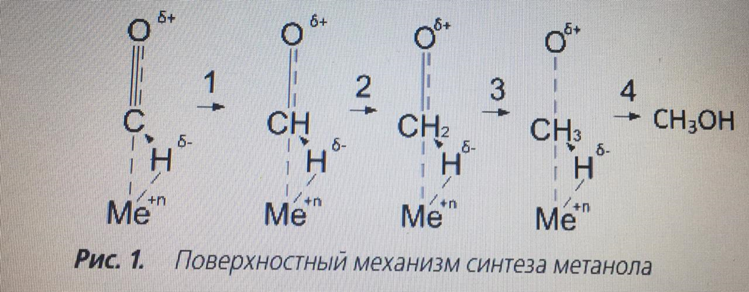
The second approach is based on the fact that the main substance of synthesis is a CO molecule, which, adsorbed on the active center of a weakly reduced catalyst, forms a positively charged chemisorbed me–CO complex [5, 6, 10]. The diffusion of hydrogen atoms under the influence of high pressure increases, so that they "dissolve" in the near-surface layer of the catalyst and when interacting with its surface, they acquire an effective negative charge. In the course of successive stages (Fig. 1) the Me–CO bond is saturated with negatively charged hydrogen atoms, resulting in loosening of this bond and splitting off of the final product – methanol. The main side reaction is the formation of hydrocarbons by the Fischer–Tropsch reaction on the metal centers of the Zn-Cu-Al catalyst that dynamically arise during operation, which in the industrial synthesis of methanol are continuously passivated by CO2 present in the initial synthesis gas at a concentration of about 8 vol.%.
Appropriate laboratory studies were conducted to confirm the proposed mechanisms. Experiments with a labeled oxygen atom [7] have shown that the source of the oxygen atom in the methanol molecule is carbon dioxide or water. At the same time, spectroscopic analyses indicate the formation of a positively charged chemisorbed Me–CO complex [5]. This paper analyzes the mechanism that takes into account the formation of surface compounds on the catalyst. This mechanism allows us to take into account the physical and chemical properties of the surface of the catalyst and most accurately predict its industrial operation. Researchers do not have a clear idea about the structure of the active center of the catalyst. Based on EPR studies, the authors of [8] indicate that the structure of the catalyst contains fragments of Cu2+ - O-M–O-Cu2+, which are probably the active centers of synthesis. Some authors [6] talk about the introduction of copper atoms into the crystal lattice of zinc oxide. To clarify the structure of the active center and substantiate the chosen mechanism, we performed confirming quantum chemical calculations. The DFT – Density Functional Study method was chosen as the calculation method for this work [9]. The theoretical approximation was the B3LYP model, the theory of the Becke density functional (B3), using the electronic correlation of Li Yang and Pair (LYP). This model is suitable for calculations of heterogeneous systems, and allows you to calculate the necessary energy parameters with sufficient accuracy. The set 6–311G** was chosen as the basis [10]. The main criterion confirming the existence of this surface structure was chosen as the binding energy between the active center of the catalyst and the adsorbed carbon monoxide molecule. This energy was calculated from the difference between the total energy of the adsorbed structure and the sum of the total energies of the CO molecule and the active center:
ERU=EClst–CO - (EClst+ECO),

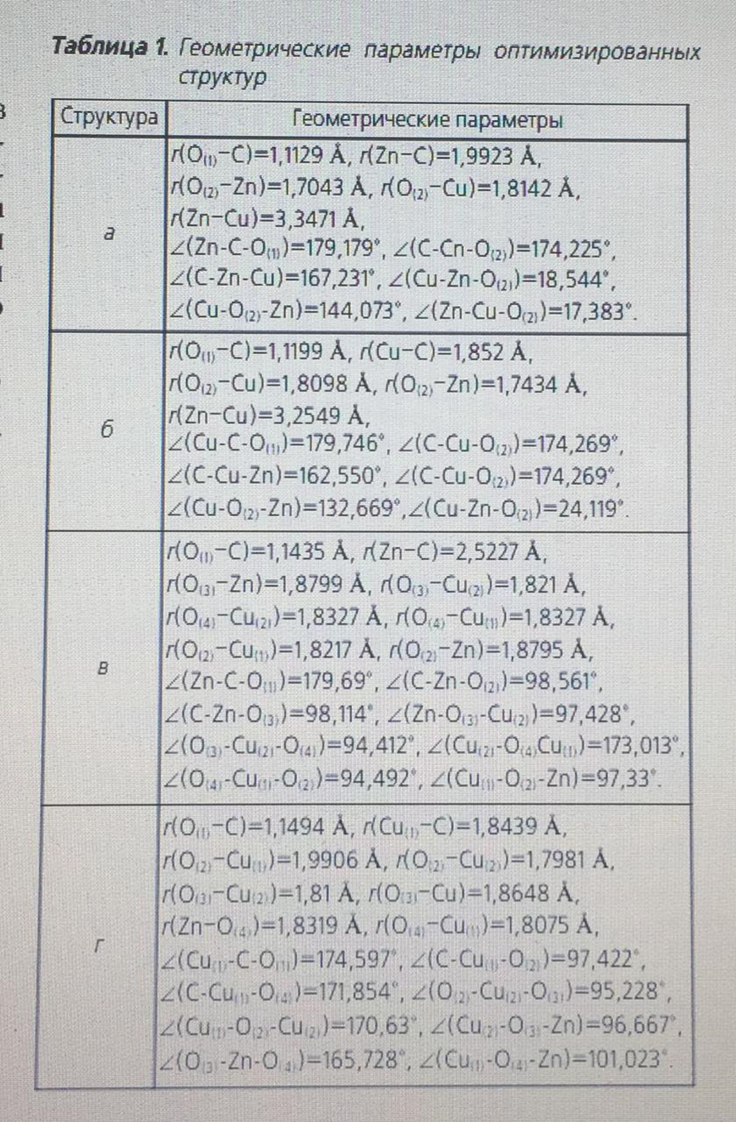
where ECB is the binding energy between the cluster and the adsorbed CO molecule; EClst-CO is the total energy of the cluster with the adsorbed CO molecule; EClst is the total energy of the cluster; ECO IS the total energy of the CO molecule. The calculation was performed for a temperature of 543 K and a pressure of 6 MPa, since these values are the average for the methanol synthesis reaction. The optimized structures shown in Fig. 2. Geometric parameters of optimized structures are shown in table. 1. in the course of optimization, no imaginary oscillation frequency was obtained, which proves their stationarity.
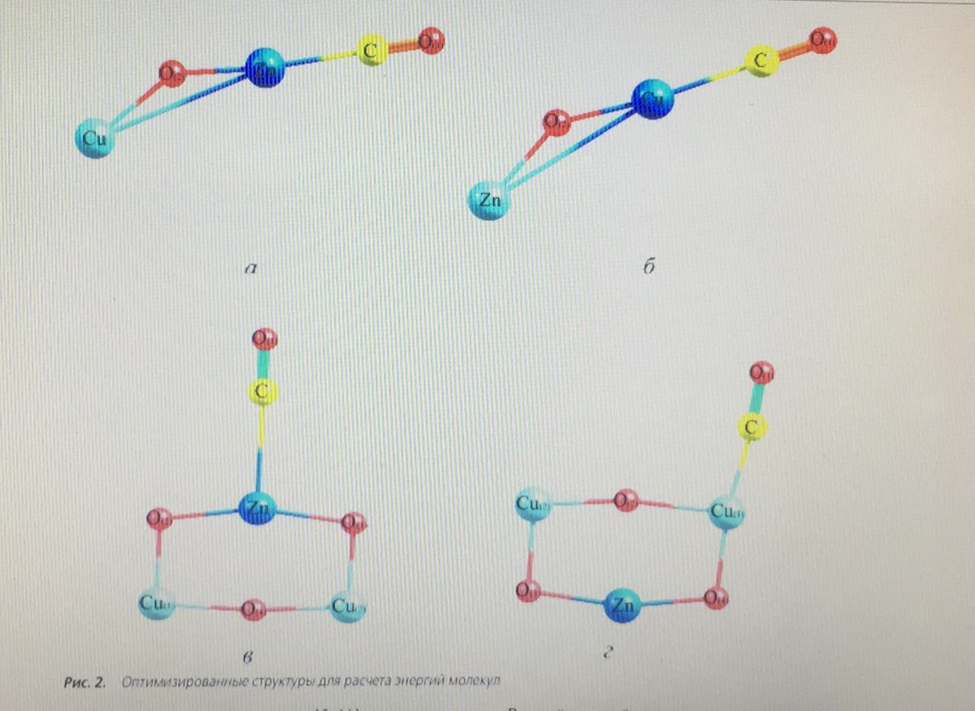
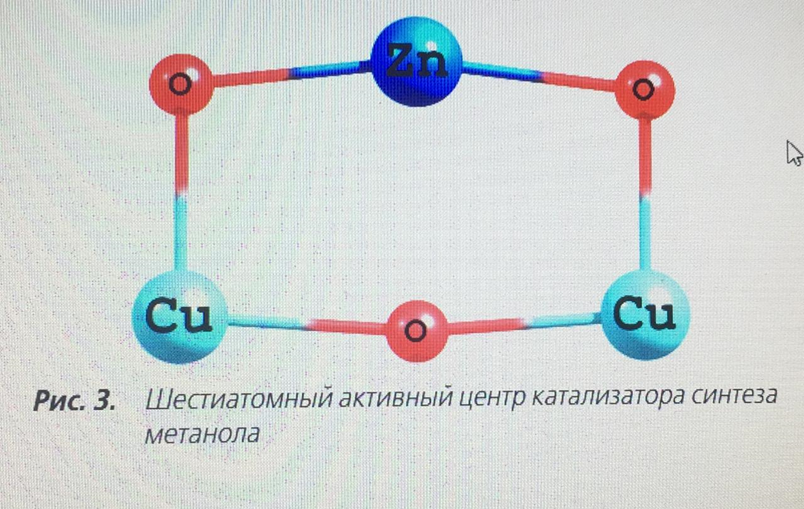
In the periodic literature [5, 11], there are opposite opinions about which of the metal atoms adsorbs the CO molecule, so the cluster energy during CO adsorption was calculated for both the zinc atom and the copper atom. Simultaneous adsorption of a CO molecule on each metal is not possible energetically, and therefore this form was not taken into account. Also in the literature there is a mention of the structure of the active center consisting of six atoms [8], i.e. of the structure Cu2+ - O-M–O-Cu2+. Based on this view, the form of the active center of the catalyst is proposed, Fig. 3.
An important task was to find out the possibility of adsorption of the CO molecule on both the zinc atom and the copper atom. The team of authors [11] speaks about the adsorption of CO molecules on individual zinc or copper atoms, but this statement is doubtful, since the probability of complete destruction of the crystal lattice of the oxide catalyst is extremely small. To calculate the binding energy of an adsorbed CO molecule, the energy of a separate CO molecule and a free active center was calculated at the same pressure and temperature values, including using the calculation method used for adsorbed forms, table. 2. as can be seen from table 1, the strongest bond is a triatomic cluster with a CO molecule adsorbed on copper (Fig. 2, b) – 308.46 kJ/mol. If the active metal is a zinc atom (Fig. 2, a), the binding energy decreases by 25.1 kJ/mol – a value sufficient to state that the active metal is a copper atom. The binding energy of the adsorbed CO molecule on the surface of hexatomic clusters (Fig. 2, b, d) is more than 2 times less, and therefore it can be argued that these structures are energetically unprofitable.

Thus, in the course of this study, a confirmatory calculation of the adsorption of a CO molecule on a low-temperature methanol synthesis catalyst was performed using quantum chemical methods. During the calculation, it was shown that the most likely active center of the catalyst is a three-atom cluster containing copper, zinc and oxygen atoms (Fig. 2, b). The most likely active metal in the cluster is a copper atom, since the bond of the CO molecule adsorbed on copper is greater than the rest. This does not exclude the possibility of adsorption of the CO molecule on zinc, since the energy of this bond is quite large – 283.36 kJ/mol, but still less than in the case of adsorption on copper. Therefore, a three-atom cluster with CO molecule adsorption on copper was chosen for the mathematical model.
Conclusions: the main approaches to consideration of CO adsorption on low-temperature Zn-Cu-Al-catalytic systems of methanol synthesis are Analyzed. The synthesis mechanism based on the formation of a positively charged chemosorbed complex on the surface of the catalyst was analyzed using quantum chemical calculation methods. The m ost frequently encountered structures in the literature, consisting of three and six atoms, were selected as the active center of the catalyst. Based on the values of binding energies obtained during the calculation, it is shown that the most energetically advantageous is the triatomic active center, whose binding energy is 308.46 kJ/mol.
LIST OF REFERENCES
1. Sekunova M. V. Methanol-an important raw material resource for the production of fuels / / Mir petrochemicals. - 2007. - № 4. - P. 4-8.
2. Karpov S. A., Kunashev L. H., Mortikov E. S., Kapustin V. M. Production of methanol: the current state of industry and development trends / / oil Refining and petrochemistry. - 2009. - № 7. - P. 3-8.
3. Slinko M. G. Modeling of chemical reactors. Novosibirsk: Nauka, 1968, 96 p.
4. Kravtsov A.V., Novikov A. A., Koval P. I. Computer analysis of technological processes. Novosibirsk: Nauka, 1988, 216 p.
5. Kravtsov A.V. on the dynamic features of the reaction mechanism of carbon monoxide hydrogenation // Questions of kinetics and catalysis. Interuniversity collection. Ivanovo: Nauka, 1980, Pp. 33-40.
6. Weigel J., Koeppel R., Baiker A. Surface species in CO and CO2 hydrogenation over copper/zirconia: on the methanol synthesis mechanism / / Langmuir. - 1996. - no. 12. - P. 5319-5329.
7. Takeuchl A., Katzer J. Mechanism of methanol formation / / Jour nal of Physical Chemistry. - 1981. - V. 52. - No. 85. - P. 937-939.
8. Altynnikov A. A., Anufrienko V. F., Rozovsky A. Ya. Detection of copper ion clusters in cu-Zn-Al oxide catalysts for methanol synthesis according to EPR data / / Kinetics and catalysis. - 1999. - Vol. 40. - No. 1. - Pp. 129-133.
9. Gokhale A. A., Kandoi S., Greely J. P., Mavrikakis M., Dumesic J. A. Molecular-level description of surface chemistry in kinetic models using density functional theory // Chemical Engineering Science. – 2004. – V. 59. – № 22-23. – P. 4679-4691.
10. Polishchuk O. Kh., Kizhner D. M. Chemical research by methods of calculating the electronic structure of molecules. - Tomsk: Izdvo TPU, 2006. - 146 p.
11. HyeWon Lim, MyungJune Park, SukHwan Kang, HoJeong Chae, Jong Wook Bae, KiWon Jun. Modeling of the Kinetics for Methanol Synthesis using Cu/ZnO/Al2O3/ZrO2 Catalyst: Influence of Carbon Dioxide during Hydrogenation // Ind. Eng. Chem. Res. – 2009. – V. 23. – № 48. – P. 10448–10455.
Жариялаған:
Талғатова Айсұлу Даулетқызы 13 Желтоқсан 2020 506
Материал ұнаса әріптестеріңізбен бөлісіңіз

Сайтқа 5 материал жариялап, тегін АЛҒЫС ХАТ алыңыз!

Сайтқа 25 материал жариялап, тегін ҚҰРМЕТ ГРОМАТАСЫН алыңыз!